
Applying Machine Learning to…..Yeast?
April 29, 2020
Posted by Ted Baltz, Senior Staff Software Engineer, Google Research, Accelerated Science Team
Humans have a long history with yeast, tied to the beginnings of plant domestication — baker’s (or brewer’s) yeast, Saccharomyces cerevisiae, has been used to make grains more digestible in the form of bread (or beer) for millennia. Today, yeast still has a large impact, with biologists adopting it as a model organism for biological research, genetics in particular, because it is easy to grow in the lab and is a eukaryote (i.e., unlike bacteria, it has a cell nucleus, like our cells do). It has even earned its own catchphrase in the biological community — “the awesome power of yeast genetics”. Studying the fundamentals of genetics is much easier in yeast, but is still applicable to humans since ~1000 yeast genes have a sequence homolog to human ones. Understanding how genes work together as a system is core to understanding all living things, which drives interest in this microorganism.
In collaboration with Calico Life Sciences, we present “Learning causal networks using inducible transcription factors and transcriptome-wide time series”, published in Molecular Systems Biology. Based on exhaustive experiments, we built a genome-wide model for the regulation of gene expression in S. cerevisiae and verified some of the results experimentally, enabling future investigations into less well understood biological systems. The Induction Dynamics gene Expression Atlas is available from Calico in a format easy to manipulate in python, with open-sourced code to do this on the Google Research GitHub. The data is hosted in a standard format at the Gene Expression Omnibus.
Using Yeast to Provide Insight into Aging
Yeast reproduce through a process called budding, in which a small bud grows from the surface of the parent to produce an offspring that is almost genetically identical. Interestingly, even though yeast are single-celled organisms, they grow old and die, typically after 30 budding events. In fact the “scars” from budding are clearly visible under a powerful microscope, allowing one to tell the age of the cell simply by looking! The problem is that researchers still do not know what causes aging to happen.
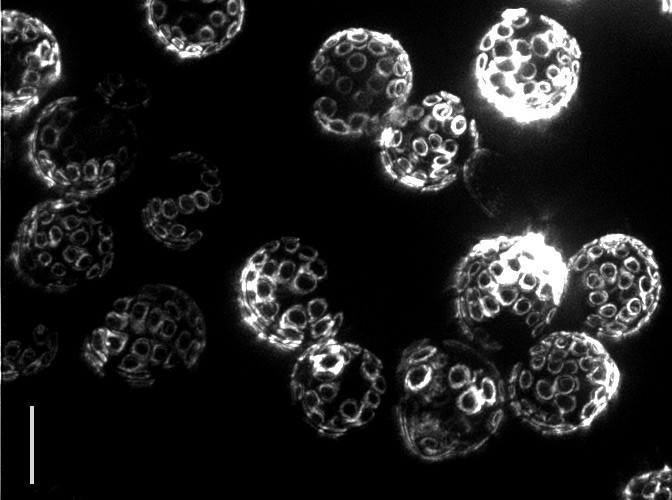 |
| Bud scars on old yeast cells (5 μm bar for scale) — Photo Credit: Ian Foe, (Calico) |
The Gene Expression Experiment
Genes encoded in DNA only function after being transcribed to RNA. It’s the RNA that is “translated” or “read” by ribosomes to produce protein. The level of protein production is governed by how much RNA is transcribed from DNA. Most of the work in a cell is being done by proteins, so they are key to understanding cell behavior. Yet, while we’d really like to measure the protein production levels, techniques to identify proteins at this scale are prohibitively expensive. Instead, in this experiment we use RNA as a proxy, since measuring RNA levels is easier.
The gene expression experiment is designed to perturb individual genes and measure, over time, how every other gene in the genome responds. The ability to rapidly perturb and track dynamics allows us to learn causal relationships and non-linear behaviors missing in most experiments. These dynamic data can also be used to train predictive models. This is made possible by strains of yeast with a single gene that is responsive to an external switch, in this case the hormone β-estradiol. To perturb a gene, the hormone is introduced, causing the switched gene to be overexpressed by a factor of 50 within 10 minutes. The yeast culture is then sampled at several points in time to measure the gene expression levels on microarrays. These experiments were done in parallel, with one yeast strain per culture, running concurrently.
Most of the perturbation experiments were done on a particular class of genes coding for transcription factors (TFs). These genes are the primary regulators of gene expression, coding for proteins that actually bind to the DNA strands, permitting or blocking transcription of particular genes.
When gene “a” is turned on it may upregulate gene “b” and downregulate gene “c”, and later lead to upregulation of gene “d”. Since yeast has more than 6000 genes, tracing the downstream impact of perturbing a single gene can get complicated very quickly. By combining experiments on different genes, one hopes to disambiguate the exact regulation mechanisms.
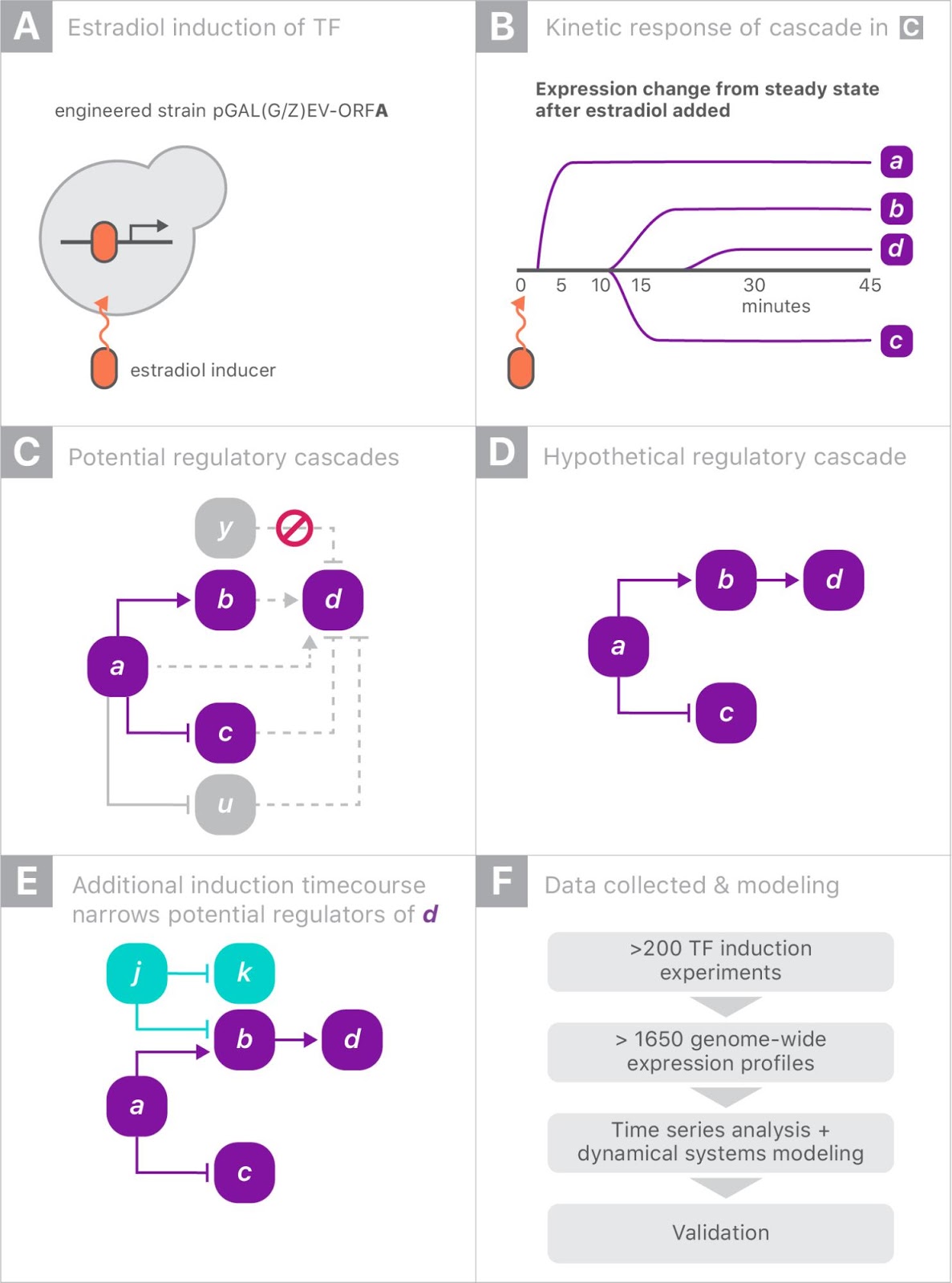 |
| Schematic of the genome perturbation experiment: yeast strain with switchable gene “a”. Turning on a single gene (A) can result in differing levels of gene expression over time (B). Tracking these changes in comparison to those induced by turning on other genes (C and D) can provide insight into the regulation mechanisms (E). |
For this experiment, we partnered with Calico because of the scale of the data, and the opportunity to leverage Google’s machine learning expertise and compute resources. There were more than 200 perturbation experiments on different yeast strains, each activating a single gene. In each experiment, the expression levels of all 6000 genes were measured eight times over 90 minutes, yielding a total of almost 20 million individual measurements (panel F, above). Clearly some automation was required to analyze the data.
Our approach was to model the whole process as a system of differential equations: the rate of change of the expression of a gene was proportional to a weighted sum of the expression levels of all genes. We first estimated the time derivatives from the data by simply subtracting the expression levels among adjacent time points. We then predicted the time derivatives using only the raw expression levels themselves. By fitting a linear regression, we are, in effect, fitting the coefficients of a system of differential equations describing gene regulation. Our hope is that the differential equation model would be a low dimensional representation of the data that could be interpreted more easily. To handle overfitting, we regularized the model using the L1-norm, which prefers to set uninformative parameters to exactly zero.
Because each of the 200 experiments was unique, we held out each one in turn, refitting the model and allowing the selection of the best hyperparameters to optimize the out-of-sample loss. In the end, the work required a significant amount of compute, amounting to more than 50 million full regularization paths.
Model Results
Our model made predictions about which genes would code for intermediate regulators of gene expression. This is an attempt at modeling the full gene regulation network of the organism. To verify these predictions, our collaborators at Calico collected more data from ten new strains of yeast. Three out of ten of the predictions held up in these experiments. One of the genes that the model predicted to be active encoded an unverified transcription factor, while another previously identified as a regulator but never followed up, was found by our model to be a very active regulator. Our model was able to identify these without prior biological knowledge, demonstrating that these ML techniques might scale to other domains or organisms that are much less well studied.
More discussion of the impact of this work within the broad context of the field of genomics is available in an independent peer commentary.
Acknowledgements
We wish to thank Marc Coram, Minjie Fan, and Marc Berndl for their foundational contributions to this work, the Google Accelerated Science team for their continual support, and the entire team at Calico for the opportunity to collaborate on this experiment.
Other posts of interest
-
April 23, 2024
Safely repairing broken builds with ML- Machine Intelligence ·
- Software Systems & Engineering
-
April 12, 2024
Contrastive neural audio separation- Machine Intelligence ·
- Sound & Accoustics
-
April 11, 2024
Patchscopes: A unifying framework for inspecting hidden representations of language models- Machine Intelligence ·
- Natural Language Processing ·
- Responsible AI